Alternative Lengthening of Telomeres: Mechanisms & Cancer
Every time a human cell divides, it loses a small piece of its chromosomal ends — the telomeres. These protective caps, built from repetitive TTAGGG sequences, act like the plastic tips on shoelaces: once they’re gone, the whole structure unravels. Normal cells tolerate only a finite number of divisions before critically short telomeres trigger senescence or programmed death.
Cancer cells cheat this clock through two strategies. Roughly 85–90% reactivate telomerase, the reverse transcriptase that rebuilds telomeric DNA enzymatically. The remaining 10–15% use something fundamentally different: alternative lengthening of telomeres (ALT), a recombination-based mechanism that copies telomeric sequences from existing DNA templates — no telomerase required.
ALT drives some of the most treatment-resistant tumors in oncology — osteosarcoma, glioblastoma, liposarcoma, high-risk neuroblastoma. For researchers hunting new drug targets and clinicians managing patients whose tumors shrug off standard therapy, ALT biology has become one of the most consequential frontiers in cancer science.
What Is Alternative Lengthening of Telomeres?
Alternative lengthening of telomeres is a recombination-based telomere maintenance mechanism used by roughly 10–15% of human cancers. First characterized in the 1990s by Bryan et al. studying telomerase-negative immortal cell lines, ALT operates through homologous recombination rather than enzymatic synthesis — making it biologically distinct and a separate therapeutic target (Bryan TM et al., Nature Medicine, 1997).
Telomeres and the End-Replication Problem
Telomeres are repetitive nucleotide sequences capping the ends of human chromosomes, protecting them from degradation and end-to-end fusion. Every round of DNA replication leaves them slightly shorter — a structural consequence known as the end-replication problem, arising because DNA polymerase cannot fully copy the lagging strand’s terminus.
Once telomeres shorten past a critical threshold, they trigger either replicative senescence or apoptosis. Cancer cells must override this limit to achieve immortality, and ALT provides the less common but equally effective route to doing so.
How ALT Works: Recombination Over Replication
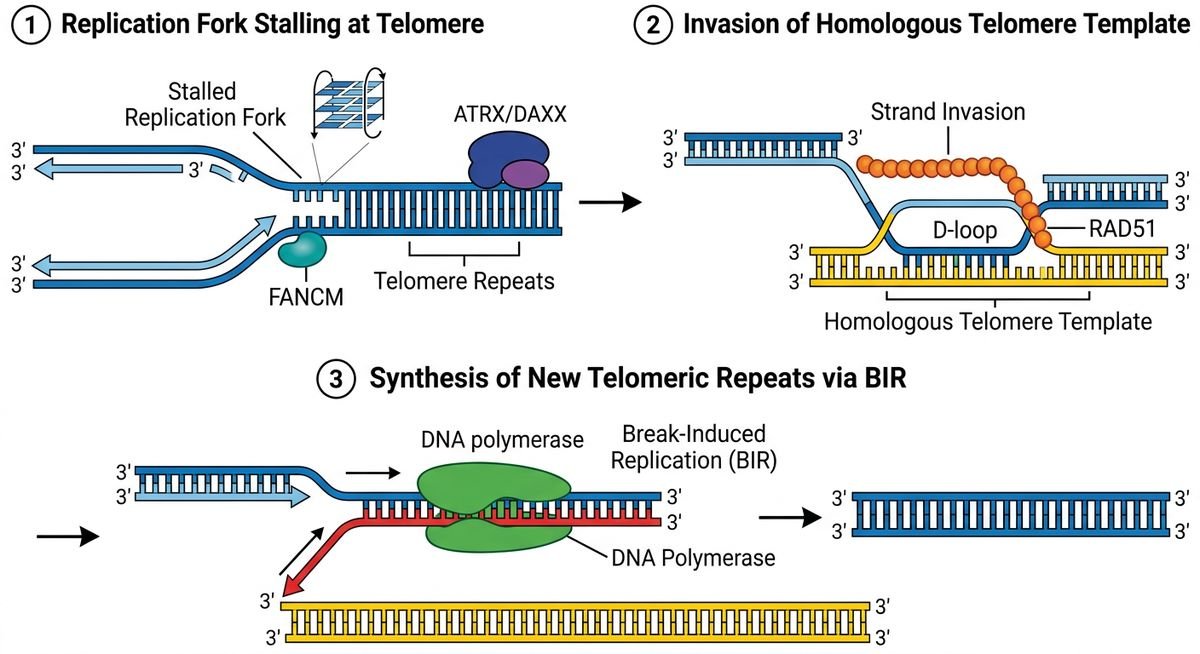
Rather than synthesizing new telomeric repeats enzymatically, ALT-positive cells use existing telomeric sequences — on sister chromatids, other chromosomes, or extrachromosomal DNA fragments — as copy templates. The dominant pathway is break-induced replication (BIR), in which a stalled or broken replication fork at a telomere invades a homologous telomeric sequence and uses it to prime long-tract DNA synthesis.
A second amplification route involves rolling-circle replication of extrachromosomal telomeric repeats (ECTRs) — circular DNA molecules composed entirely of telomeric sequence. These ECTRs serve as mobile templates that can dramatically and rapidly extend individual chromosome ends, producing the strikingly heterogeneous telomere lengths that define ALT-positive tumors.
BIR-mediated ALT synthesis is semi-conservative and highly mutagenic compared to normal replication, introducing genomic instability that may further drive tumor evolution (Dilley RL et al., Nature Structural & Molecular Biology, 2016).
| Feature | Telomerase-Based Maintenance | ALT |
|---|---|---|
| Core mechanism | Enzymatic reverse transcription (TERT + TERC) | Homologous recombination / BIR |
| Telomere length pattern | Relatively uniform | Highly heterogeneous (short to very long) |
| Cancer prevalence | ~85–90% of human cancers | ~10–15% of human cancers |
| Key genetic driver | TERT promoter mutations or amplification | ATRX/DAXX loss-of-function mutations |
| Primary cancer types | Epithelial carcinomas (breast, lung, colon) | Sarcomas, gliomas, neuroendocrine tumors |
| Diagnostic biomarker | TRAP assay (telomerase activity) | C-circle assay, APB detection |
Molecular Hallmarks and Key Proteins Involved in ALT
ALT-positive cells carry four canonical molecular signatures that distinguish them from telomerase-driven tumors: ALT-associated PML bodies (APBs), C-circles, heterogeneous telomere length, and elevated telomere sister-chromatid exchange (T-SCE). Loss-of-function mutations in the ATRX/DAXX chromatin-remodeling complex are the single most frequent genetic event enabling this phenotype.
The ALT Phenotype: What Makes a Cell ALT-Positive
The most visually striking hallmark is the ALT-associated PML body, or APB — nuclear foci where telomeric DNA, telomere-binding proteins, and promyelocytic leukemia (PML) protein co-localize. These are essentially recombination hubs assembled at chromosome ends, and their presence alone is considered strong presumptive evidence of ALT activity.
C-circles are partially single-stranded, extrachromosomal circular DNA molecules composed entirely of telomeric repeats. They are exquisitely specific to ALT cells and detectable in patient blood samples, making them one of the most diagnostically useful biomarkers in the field. C-circle abundance correlates directly with ALT activity levels across cell lines (Henson JD et al., Proceedings of the National Academy of Sciences, 2009).
Heterogeneous telomere length is a third defining feature. Unlike telomerase-maintained cells, which tend toward uniform telomere lengths, ALT cells display wildly variable ends — some extremely short, some extraordinarily long — within the same nucleus. Elevated T-SCE rounds out the quartet, reflecting the high rate of recombination between sister chromatids at telomeric loci.
ATRX and DAXX: Gatekeepers Lost in ALT Cancers
The ATRX (Alpha-Thalassemia/Mental Retardation X-linked) protein and its chaperone partner DAXX normally deposit histone H3.3 at telomeric and pericentromeric heterochromatin, maintaining a tightly compacted, recombination-resistant chromatin state. When ATRX is mutated or silenced — which occurs in roughly 90% of ALT-positive gliomas and a substantial proportion of ALT-positive sarcomas — that repressive architecture collapses.
The result is a permissive chromatin environment where telomeric sequences become accessible to the homologous recombination machinery. ATRX loss also allows TERRA (telomeric repeat-containing RNA) to accumulate, destabilizing telomeric heterochromatin and promoting R-loop formation (Lovejoy CA et al., PLoS Genetics, 2012). Critically, ATRX mutation alone is not sufficient to activate ALT — but it removes a major barrier.
| Protein | Normal Function | Effect of Loss/Mutation in ALT |
|---|---|---|
| ATRX | Deposits H3.3 at telomeric heterochromatin | Destabilizes telomeric chromatin; permits aberrant HR |
| DAXX | Histone H3.3 chaperone; partners with ATRX | Loss phenocopies ATRX mutation; common in PanNETs |
| FANCM | Suppresses BIR at stalled replication forks | Loss increases BIR-dependent ALT activity |
| RAD51/RAD52 | Mediate strand invasion in HR | Essential for ALT recombination; potential drug targets |
| SLX4 complex | Resolves recombination intermediates | Required for processing ALT-associated HR structures |
Other Critical Regulators
FANCM (Fanconi Anemia Complementation Group M) acts as a natural brake on ALT. It unwinds branched DNA intermediates at stalled replication forks, suppressing the strand invasion events that drive BIR-based telomere extension. When FANCM is depleted experimentally, ALT activity surges — making it both a mechanistic gatekeeper and a potential therapeutic lever (Pan X et al., Nature Communications, 2019).
RAD51 and RAD52 mediate the strand-invasion step that initiates template-directed telomere synthesis. TERRA, the long non-coding RNA transcribed from telomeric sequences, accumulates at ALT telomeres and promotes R-loop formation — RNA:DNA hybrid structures that further expose single-stranded telomeric DNA to the recombination machinery.
Which Cancers Use ALT? Prevalence Across Tumor Types
ALT clusters heavily in tumors of mesenchymal and neural origin. The pattern is not random — tissues with low baseline telomerase activity during normal development are more permissive for ALT selection during oncogenesis, while epithelial cancers overwhelmingly favor telomerase reactivation.
High-Prevalence ALT Cancers
Osteosarcoma sits near the top of every prevalence estimate, with approximately 50–60% of tumors maintaining telomeres through ALT. Liposarcoma follows closely, particularly the dedifferentiated subtype, at roughly 50%. Both are mesenchymal tumors with characteristically high rates of ATRX mutation.
Gliomas present a more nuanced picture. In IDH-mutant, lower-grade astrocytomas, ALT prevalence climbs to 60–65%, largely because IDH mutation and ATRX loss co-occur at high frequency. Glioblastoma (IDH-wildtype) shows lower but still clinically meaningful ALT rates of around 25–30%.
Neuroblastoma deserves special mention. ALT-positive neuroblastoma represents a minority of cases, but it defines the highest-risk biological subgroup — associated with older age at diagnosis, absence of MYCN amplification, and markedly worse survival compared to both telomerase-positive and telomere-stable disease (Dagg RA et al., Cancer Research, 2017).
| Cancer Type | ALT Prevalence | Key Genetic Co-alteration |
|---|---|---|
| Osteosarcoma | ~50–60% | ATRX mutation/loss |
| Liposarcoma (dedifferentiated) | ~50% | ATRX mutation/loss |
| IDH-mutant astrocytoma (LGG) | ~60–65% | IDH1/2 mutation + ATRX loss |
| Glioblastoma (IDH-wildtype) | ~25–30% | ATRX mutation (subset) |
| Neuroblastoma (high-risk subset) | ~25% of high-risk cases | ATRX/DAXX mutation; no MYCN amplification |
| Pancreatic neuroendocrine tumors | ~60% | ATRX or DAXX mutation |
Prognostic Significance
ALT status carries prognostic weight that varies by tumor type. In neuroblastoma, ALT positivity is an independent marker of poor survival. In pancreatic neuroendocrine tumors, ATRX/DAXX loss — which serves as a reliable proxy for ALT — predicts more aggressive behavior and higher metastatic potential. In IDH-mutant gliomas, the prognostic picture is more complex: ATRX loss defines the astrocytoma molecular subtype but does not uniformly predict worse outcomes within that category.
Detecting ALT: Biomarkers and Diagnostic Methods
No single assay definitively confirms ALT status, but several complementary approaches are used in both research and clinical settings. The most widely deployed is the C-circle assay — a rolling-circle amplification-based test that quantifies extrachromosomal telomeric circles with high specificity for ALT-positive cells.
APB detection via immunofluorescence (co-localizing PML protein with telomeric FISH probes) provides visual confirmation of ALT activity at the single-cell level. Telomere-length heterogeneity assessed by terminal restriction fragment (TRF) analysis or telomere FISH shows the characteristic broad distribution unique to ALT cells.
For clinical pathology, immunohistochemistry for ATRX protein expression offers a practical surrogate. Loss of nuclear ATRX staining strongly correlates with ALT positivity and is now routinely included in the diagnostic workup for gliomas and pancreatic neuroendocrine tumors (Heaphy CM et al., Science, 2011).
Therapeutic Targeting of ALT
ALT-positive cancers are inherently resistant to telomerase inhibitors like imetelstat — they simply don’t use the target enzyme. This has driven intense interest in therapies that exploit ALT-specific vulnerabilities.
ATR Inhibitors
ALT cells generate high levels of replication stress at telomeres, making them dependent on the ATR (Ataxia Telangiectasia and Rad3-related) kinase for DNA damage tolerance. ATR inhibitors such as elimusertib (BAY 1895344) and ceralasertib (AZD6738) have shown selective toxicity against ALT-positive cell lines in preclinical studies, with several now in clinical trials for ATRX-mutant tumors (Flynn RL et al., Science, 2015).
Targeting the Recombination Machinery
Disrupting the HR proteins that ALT depends on is another active strategy. FANCM inhibition amplifies replication stress specifically at ALT telomeres, creating a synthetic lethal vulnerability. Small-molecule FANCM inhibitors are in early preclinical development. PARP inhibitors, already approved for BRCA-mutant cancers, show enhanced activity in some ALT-positive contexts by further destabilizing the replication fork repair pathways these cells rely on.
Combination Approaches
Emerging data suggest that combining ATR inhibition with either PARP inhibitors or conventional DNA-damaging agents may overcome resistance mechanisms in ALT tumors. The underlying logic is straightforward: ALT cells already operate near the ceiling of tolerable replication stress, and pushing them past that threshold triggers mitotic catastrophe or apoptosis.
Frequently Asked Questions
What is the difference between ALT and telomerase?
Telomerase is an enzyme (a reverse transcriptase) that adds telomeric repeats to chromosome ends using an RNA template. ALT achieves the same outcome — telomere extension — but through homologous recombination, copying sequences from other chromosomes or extrachromosomal DNA. About 85–90% of cancers use telomerase; 10–15% use ALT. The two mechanisms are largely mutually exclusive within individual tumors.
Can a cancer cell use both ALT and telomerase simultaneously?
Rare cases of co-occurrence have been documented, but they are the exception. Most tumors commit to one pathway. Some evidence suggests that individual cancer cells may switch between mechanisms under selective pressure — for instance, when telomerase is pharmacologically inhibited — though this remains an active area of investigation.
How is ALT detected in a tumor sample?
The gold-standard combination includes the C-circle assay (highly specific for ALT), immunofluorescence for APBs, and assessment of telomere length heterogeneity via FISH or TRF analysis. In clinical practice, loss of ATRX nuclear expression on immunohistochemistry serves as a reliable surrogate marker, particularly for gliomas and pancreatic neuroendocrine tumors.
Why does ALT matter for cancer treatment?
ALT-positive tumors are inherently resistant to telomerase-targeting drugs like imetelstat, which eliminates an entire class of therapy. However, ALT creates its own vulnerabilities — particularly dependence on the ATR-mediated DNA damage response — that are now being exploited by drugs in clinical trials, including ATR inhibitors and PARP inhibitors.
Which cancers are most likely to be ALT-positive?
ALT is most prevalent in tumors of mesenchymal and neural origin: osteosarcoma (50–60%), dedifferentiated liposarcoma (~50%), pancreatic neuroendocrine tumors (~60%), and IDH-mutant astrocytomas (60–65%). It is rare in common epithelial cancers such as breast, lung, and colorectal carcinoma.
Is ALT inherited or does it develop during cancer progression?
ALT is an acquired phenotype that emerges during tumor evolution, not an inherited trait. It typically arises when cancer cells lose ATRX or DAXX function through somatic mutation, creating the permissive chromatin environment needed for recombination-based telomere maintenance. Germline ATRX mutations (as seen in ATR-X syndrome) do not cause ALT activation in normal tissues.
The Road Ahead
Alternative lengthening of telomeres has evolved from a molecular curiosity into a clinical priority. The convergence of improved diagnostic tools — particularly the C-circle assay and routine ATRX immunohistochemistry — with ALT-targeting drug candidates like ATR inhibitors means that identifying ALT status in a patient’s tumor now has direct therapeutic implications.
The next five years will likely determine whether ALT-specific therapies can match the success of telomerase inhibitors in clinical trials. For the 10–15% of cancer patients whose tumors rely on recombination rather than telomerase, those answers cannot come soon enough.
Last modified: March 19, 2026

